Vaskulitssjukdomar: Klassificering och immunsystem av vaskulit
Vaskulit är en beskrivande term associerad med en heterogen grupp av sjukdomar som resulterar i inflammation i blodkärl.
Arterier och vener av någon storlek i något organ kan påverkas och leda till ischemiska förändringar i organet. Orsakerna eller mekanismerna bakom vaskulär inflammation är inte tydligt kända. Det viktigaste diagnostiska testet av vaskulit är ofta biopsin hos det drabbade organet. Det fullständiga blodtalet visar ofta egenskaper hos kronisk anemi tillsammans med trombocytos. Ofta finns det lymfoperu.
Henoch-Schonlein purpura och Kawasakis sjukdom är barnets vanligaste vaskulider. Det är inte så lätt att diagnostisera vaskulit. Särskilda kriterier har lagts för många vaskulitsyndrom. Men hos en patient som inte uppfyller alla kriterier är diagnosen inte lätt. Vanligtvis förekommer patienter med vaskulit med nonspecifika konstitutionella symtom som feber, myalgi, anorexi och viktminskning. Diagnos får inte göras förrän mer specifikt organs deltagande inträffar.
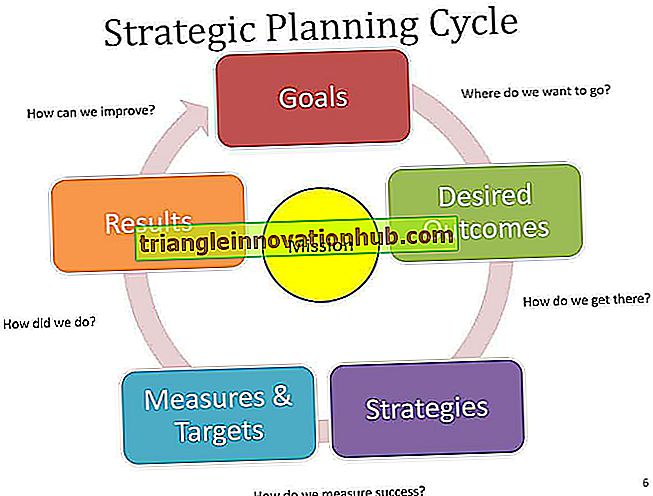
Klassificering av vaskulit:
Klassificeringen av vaskulit är svår och utvecklas fortfarande. Många klassificeringar har föreslagits om storleken på det aktuella fartyget. Den internationella konsensuskonferensen 1994 i Chapel Hill, North Carolina, har föreslagit följande klassificeringsschema för vascuhtis.
jag. Storskalig kärlvaskulit:
1. Temporal arterit:
Granulomatös arterit hos aorta och huvudgrenar, särskilt de extra kraniala grenarna i halshinnan som vanligtvis förekommer hos patienter äldre än 50 år.
2. Takayasus arterit:
Granulomatös arterit hos aorta och huvudgrenar som vanligen förekommer hos patienter yngre än 50 år.
ii. Medelstor kärlvaskulit:
1. Polyarterit nodosa:
Nekrotiserande vaskulit hos medelstora eller småstora artärer utan involvering av stora artärer, vener eller venules; njursamverkan utan glomerulonefrit.
2. Kawasakis sjukdom:
Medel- och småstimulär arterit av barndom i samband med mukokutant lymfkörtelsyndrom; mest vanliga på kranskärlen, även om vener och aorta kan vara involverade. (Lesioner av aorta har hittats vid obduktion.)
III. Vasculit i liten storlek:
1. Wegeners granulomatos:
Granulomatös inflammation av små till medelstora kärl som involverar luftvägarna; nekrotiserande glomerulonephritis vanliga.
2. Churg-Strauss syndrom:
Eosinofilrik och granulomatös inflammation som involverar andningsorganen och nekrotiserande vaskulit hos små till medelstora kärl; associerad med astma och eosinofili. (Under klassificeringen av American College of Rheumatology och traditionella klassificeringar, grupperas Wegeners granulomatos och Churg-Strauss syndrom tillsammans med polyarterit nodosa under medelstor kärlvaskulit.)
3. Mikroskopisk polyangit:
Pauci-immune nekrotiserande vaskulit som involverar små och medelstora kärl; nekrotiserande glomerulonephritis vanliga; pulmonell capillarit ofta.
4. Henoch-Schonlein purpura:
Vasculit med små kärl med immunoglobulin en immunförsvar; involvering av hud, tarm och glomeruli typiskt; associerad med artralgi och artrit.
5. Väsentlig kryoglobulinemisk vaskulit:
Vasculit med cryoglobulin immunförsvar som påverkar arterioler och venoler; associerad med serumkryoglobuliner; hud och glomeruli är ofta involverade.
6. Kutan leukocytoklastisk vaskulit:
Isolerad kutan vaskulit utan systemisk vaskulit eller glomerulonefrit.
7. Eventuell tromboflebit eller ytlig venös trombos:
Som resultat av vaskulitskador med endotelialisering hos barn, oftare på grund av hyperkoagulerbara tillstånd eller kateterinstrumentering.
Immunmekanismer av vaskulit:
Följande immunmekanismer kan spela viktiga roller i vaskulitstörningarna.
1. Autoantikroppar:
Autoantikroppar återfinns i cirkulationen av några av vaskulitstörningarna. Orsakerna till utvecklingen av autoantikroppar eller de patogena rollerna, om några, spelas av autoantikropparna är inte kända.
Antiendoteliala antikroppar:
Antiendoteliala antikroppar återfinns i vaskulitpatienter. De antiendoteliella antikropparna kan inducera komplementaktivering eller ADCC (antikroppsberoende cellulär cytotoxicitet) eller intravaskulär trombos.
Antineutrofila cytoplasmiska antikroppar (ANCAs):
Antineutrofil-cytoplasmiska antikroppar (ANCAs) är närvarande i cirkulationen av vissa vaskulitstörningar. ANCAs reagerar med cytoplasmatiska antigener av neutrofiler och reaktionen kan vara ansvarig för den vaskulära inflammatoriska processen. Indirekt immunfluorescensmikroskopi (IIFM) och ELISA är de vanligaste metoderna för att detektera ANCA.
Baserat på immunofluorescensfärgningsmönstren för neutrofil-cytoplasman beskrivs två typer av ANCA: er:
en. Cytoplasmiska ANCAs (c-ANCAs)
b. Perinuclear - ANCAs (p-ANCAs)
IIFM för ANCAs utförs med användning av normala humana neutrofiler som immunofluorescenssubstratet, c- ANCA karakteriseras av fingranulär färgning av neutrofil cytoplasma med central accentuering mellan nukleärlabben; men själva kärnan fläckar inte. Proteinas 3 (PR-3), ett 29 KD neutralt serinproteas (lokaliserat i neutrofil primära granuler) är det antigena målet för c-ANCAs.
p-ANCA kännetecknas av immunofluorescensfärgning av perinuclearområdet. Antigenmålet för p-ANCAs i vaskulitstörningar är myeloperoxidas (MPO). Det perinukleära färgmönstret orsakas av omfördelningen av målantigener från cytoplasman till kärnområdet, när etanol används för att framställa humana neutrofiler som substrat för IIFM-studien . (När formalin används istället för etanol för att fixera neutrofilerna på glasskenan, mobiliseras inte målantigenen till kärnområdet, och immunfluorescensmönstret för färgning är cytoplasmatisk.)
ELISA för detektering av proteinas 3 (PR-3 ANCA; c-ANCA) och myeloperoxidas (MPO-ANCA) är kommersiellt tillgängliga. Både c-ANCA (PR-3-ANCA) och p-ANCA (MPO-ANCA) är associerade med Wegeners granulomatos, polyarterit nodosa inklusive mikroskopisk polyarterit, Churg-Strauss syndrom, idiopatisk pauciimmun-nekrotiserande och crescentrisk glomerulonephritis och överlappande polyangit syndrom.
Aktivering av neutrofiler leder till translokationen av proteinas 3 från cytoplasman till cellytan. Anti-proteinas 3-antikropparna i cirkulationen binder till proteinas 3 på neutrofila ytor och kan leda till degranulering och respiratorisk utbrott av neutrofiler, vilket resulterar i en inflammatorisk skada på vaskulära vävnader.
Det finns rapporter om association av ANCA med inflammatoriska sjukdomar i mag-tarmkanalen (såsom ulcerös kolit, Crohns sjukdom) och hepatobiliära (primära skleroserande kolangit, kronisk aktiv hepatit, primär biliär cirros). ANCA har också rapporterats vara närvarande i många andra sjukdomar som SLE, Feltys syndrom, reumatoid artrit, Sjogrens syndrom, juvenil reumatisk artrit etc. Ett positivt c-ANCA-resultat hos en individ föreslår vidare utvärdering av individen för Wegeners granulomatos.
2. Immunkomplex:
Immunkomplexmedierade reaktioner kan också vara involverade i vaskulitprocessen hos vissa vaskulitidier. Cirkulerande immunkomplex kan sätta in i kärlväggen och initiera en inflammatorisk reaktion genom att aktivera komplementproteinerna. Fc-receptorerna på makrofager och neutrofiler kan binda till Fc-regionerna av antikroppar i de vävnadsavsatta immunkomplexen och aktiveras.
De aktiverade neutrofilerna släpper ut substanser som kan orsaka inflammatorisk reaktion vid platsen för immunkomplex deponering. Ökat uttryck av vidhäftningsmolekyler på neutrofiler och endotelceller kan leda till vidhäftning av leukocyter till kärlväggen och efterföljande extravaseringar av neutrofiler i de perivaskulära områdena.
3. CD4 + T-celler, CD8 + T-celler och makrofager:
CD4 + T-celler, CD8 + T-celler och makrofager har observerats i vaskulitskadorna. De cytokiner som utsöndras av dessa celler kan bidra till den inflammatoriska processen vid vasulitidis störningar. Diagnos av vaskulit hos en individ är en svår uppgift för klinikerna.
En vaskulitpatient presenterar vanligen med icke-specifika konstitutionella fynd. Diagnos får inte göras förrän mer specifikt organs deltagande inträffar. Att göra diagnos hos en patient med vaskulit är en utvecklingsprocess, eftersom senare organsystem involvering kan leda till en omprövning av initial diagnos.
Giant cellaritit:
Giant cellartärit (GCA) eller temporal arterit (TA) är ett systemiskt, inflammatoriskt, vaskulärt syndrom som övervägande påverkar kraniala artärer. I slutet av 1800-talet rapporterade Jonathan Hutchison om en man som hade svårt att bära en hatt på grund av sina ömma tidsmässiga artärer.
Orsaken till GCA är inte känd. Sjukdomen påverkar individer under sitt sjätte årtionde. GCA är vanligtvis en självbegränsad sjukdom, med en genomsnittlig längd på cirka 2 år av sjukdomsaktivitet. De involverade kärlväggarna hos GCA-patienter infiltreras med CD4-celler av THl-typ och makrofager. De inflammerade temporala artärerna innehåller IFNy och IL-2 producerad av THl-celler. IL-la, IL-6 och TNFa producerad av makrofager finns närvarande i artärerna. Koncentrisk intimal hyperplasi är en viktig patologisk lesion i GCA.
Kliniska egenskaper:
GCA-patienter kan förekomma med feber av okänt ursprung, visuell förlust eller lemklaudikation. Patienter klagar ofta på sjukdom och trötthet.
jag. Inblandning av yttre halspulsåder orsakar huvudvärk, hårbottenvärk och tidsmässig artärartärhet. Käftklaudikation och smärta (främst i massetermusklerna under tuggning) är högspecifika symtom på 50 procent av GCA-patienter. Patienter med maxillär eller lingual arterit kan ha käke eller tunga smärta när de tuggar eller pratar. Temporala artärer är framträdande, pärlstav och öm och puls mindre. Ett normalt utseende av tidsmässiga arterier utesluter emellertid inte en diagnos av GCA.
ii. Minskad syn som är sekundär för oftalmisk arterit är den vanligaste allvarliga konsekvensen av GCA. Infarkt av den optiska nerven eller ocklusion av central retinär artär kan orsaka blindhet.
III. Aortit, särskilt av bröstkörteln är inte ovanligt och dissektion sker hos patienter med aktiv sjukdom. Thoracic aneurysm med gigantiska celler i vävnaderna kan utvecklas så sent som 15 år efter diagnosen och framgångsrik behandling av GCA.
iv. Cerebrovaskulär kärl ocklusioner kan också uppstå. Carotidartären, ryggradsartären och intrakraniella kärl kan också vara involverade. Dövhet och svimmelhet kan förekomma hos patienter med vertebralartär involvering.
Laboratorieundersökningar:
jag. Laboratoriesalarmärket för GCA är förhöjd ESR och CRP. ESR fungerar som en användbar guide till sjukdomsaktivitet och även som en grov gaze av patientens respons på behandlingen.
ii. Frekvensen av RF, ANCA och andra autoantikroppar är inte högre än för åldersmatchade kontroller.
III. Serumtransaminaser kan vara förhöjda och cirka en tredjedel av GCA-patienterna har en förhöjd alkalisk fosfatasnivå.
iv. Komplementnivåer är normala.
v. Kryoglobuliner och monoklonala immunoglobuliner är frånvarande.
vi. Histologi avslöjar en inflammatorisk infiltration, övervägande av mononukleära celler, som vanligtvis involverar hela kärlväggen (dvs panarterit).
Fibrinoidnekros är inte ett särdrag hos lesionen. Intern elastisk laminafragmentering av kärlet är en karakteristisk egenskap. Jätteceller är vanligtvis närvarande och de verkar svika delar av det inre elastiska lamina. Men frånvaron av jätteceller i vissa biopsier utesluter inte GCA. Intimal proliferation är markerad. Lesionerna av större kärl i GCA liknar de hos Takayasus arterit.
Två till tre cm av den tidsmässiga artären på symtomatisk sida tas för histologiska studier. Om en specifik del av artären är öm, pärlstav eller inflammerad bör biopsi också inkludera den delen. Eftersom lesionen är segmental i naturen undersöks flera histologiska sektioner.
Om ensidigt temporärt arteriebiopsi resultat är negativt och patienten har starkare bevis på GCA, kan kontralateral temporal arteriebiopsi utföras. Långtidsbehandling med kortikosteroider används för att behandla GCA-patienter. Vanligtvis försvinner symtomen på 36-72 timmar av kortikosteroidbehandling.
Dosen av steroid är avsmalnande under en period av månader till den lägsta dosen som behövs för att kontrollera symptomen. Inom ett år kommer de flesta patienterna att ha underhållsdoser på mindre än 10 mg qd. Det andra alternativa läkemedlet är metotrexat.
Återkallelser av GCA inträffar efter ett genomsnitt på 2 år. Sena kliniska återfall inträffar efter steroidavbrott. Den långsiktiga överlevnaden hos GCA-patienter som går in i eftergift skiljer sig inte från den normala befolkningens.
Polymyalgi Rheumatica:
Polymyalgia rheumatica (PMR) och jättecellartär arterit (GCA) är nära associerade sjukdomar och de påverkar främst äldre individer. Polymyalgia rheumatica kännetecknas av symmetrisk proximal ledning och muskelsår, ömhet och styvhet.
Dessa symptom är mest framträdande i axel-, nack- och bäcken och kan involvera distala leder och muskelgrupper. Symptomen kan uppstå plötsligt eller kan vara smutsiga över veckor till månader. Aching och styvhet är värre på morgonen och med ansträngning. Musklerna kan vara ömma. Sjukdomen kan leda till atrofi av muskler och kontraktur kan utvecklas.
PMR och GCA kan representera två delar av ett enda sjukdomsspektrum, med GCA vid den mer allvarliga änden. Båda enheterna har konstitutionella symptom. Cirka 50 procent av GCA-patienter har egenskaper hos PMR. PMA förekommer hos individer äldre än 50 år och sjukdoms etiologin är okänd.
ESR och CRP är förhöjda i PMR-patienter. Histologiska fynd i muskelbiopsi är inte diagnostiska i PMR. Temporal arteriebiopsi kan övervägas om en patient har symptom och tecken som tyder på GCA eller inte svarar för 15 mg prednisolon dagligen.
Temporal arteriebiopsi kan indikeras vid upparbetning av en äldre patient med feber av okänt ursprung med en hög ESR i vilken infektions- och malignitetstestning inte har uppnåtts. Prednisolon är valet av läkemedel för PMR. Ett snabbt och dramatiskt kliniskt svar på lågdos prednisolon (<15 mg / dag) är ett viktigt inslag i PMR.
Takayasus arterit:
Takayasus arterit (TA) är en kronisk inflammatorisk sjukdom hos stora artärer, som vanligtvis påverkar aortan och dess stora grenar och lungartärerna. Avancerade lesioner visar en panarterit med intimal proliferation. Huvudmålet för TA är hjärnan. Dr Mikito Takayasu beskrev först sjukdomen först 1905.
Etiologin hos TA är okänd. Flera etiologiska särdrag har föreslagits för att förklara inflammationsprocessen i TA, inklusive spirocheatal infektion, Mycobacterium tuberculosis infektion. Streptokockinfektioner samt cirkulerande autoantikroppar på grund av ett autoimmunt fenomen.
Den vaskulära inflammatoriska processen i TA kan leda till stenos eller ocklusion eller aneurysm hos det involverade kärlet. TA är främst en sjukdom hos unga kvinnor (särskilt de av asiatiska anor), särskilt de som är i den bärande åldern. Män påverkas sällan.
Sjukdomen TA sägs framsteg i tre steg.
jag. Den första etappen är ett tidigt systemiskt stadium. Patienten klagar över konstitutionella symtom (t ex trötthet, illamående, elakhet, feber). Den första etappen anses vara prekulitisk.
ii. Det andra steget är det vaskulära inflammatoriska skedet. Under andra etappen tenderar stenos, aneurysmer och kärlsmärta (carotidyni) att uppstå. Symptomen på vaskulär insufficiens inkluderar ängsel i armen, suddig syn, dubbel vision, stroke, övergående ischemiska attacker, hemiplegi, paraplegi och anfall.
III. Den tredje etappen är det utbrända scenen. Fibrosis sätter in, och är i allmänhet associerad med remissioner.
Huvuddetekteringen av TA är frånvaron av puls (er) eller en pulsskillnad på mer än 30 mmHg mellan höger och vänster armar. Vascular bruits hörs, oftast i carotid och bukpatienter, men också i subklaverna och femorala artärer.
en. Koronarartär involvering kan resultera i angina, myokardinfarkt, ischemisk kardiomyopati eller plötslig död.
b. Visceral kärl involvering kan orsaka buksmärtor, tarm claudication och hypertoni.
c. Retinalartärer påverkas hos en tredjedel av patienterna som orsakar retinalischemi och mikroaneurysmer.
d. Inblandning av större kärl i extremiteter kan orsaka claudicering.
e. Erythema nodosum, pyoderma gangrenosum och Raynauds fenomen är hudens manifestationer.
f. Hos gravida kvinnor kan blodtrycket inte vara mätbart på grund av pulslöshet och därför är hanteringen av graviditeten svår. Obehandlat blodtryck kan leda till subaraknoida eller intrakraniella blödningar, anfall, preeklampsi, aortisk regurgitation, synkope, fetala komplikationer och nefritisk syndrom.
Laboratorieundersökningar:
jag. Angiografi är standardkriterierna för diagnos. Angiografi avslöjar omfattande oregelbundna stenoser eller ocklusioner av aortan och dess huvudgrenar, särskilt de subklavia artärerna.
ii. Histologi av TA är oskiljaktig från jättecellerartit. De vaskulära lesionerna är initialt inflammatoriska och blir senare ocklusiva. I den tidiga fasen sker granulomatösa förändringar i mediet och adventitia av aortan och dess grenar, följt av intimal hyperplasi, medial degenerering och adventitial fibros av den sklerotiska typen. Inflammatoriska celler, övervägande, CD4 + och CD8 + T-celler, makrofager, plasmaceller och histiocyter invaderar adventitia och media men inte intima.
I vaso-ocklusivstadiet ersätts adventitierna och medierna med fibrös ärr, vasa-vasorum utplånas och intima genomgår oregelbunden förtjockning. Kortikosteroider används för att kontrollera inflammatorisk process. När remissioner med kortikosteroider inte uppnås används cytotoxiska läkemedel som metotrexat eller cyklofosfamid. Perkutan angioplastik eller bypasstransplantation kan vara nödvändig. TA är en kronisk återkommande process. 5 års överlevnadstakt för TA är 90 procent.
Polyarteritnodosa:
Polyarterit nodosa (PAN) är en nekrotiserande vaskulit av medelstora arterier (0, 5 till 1, 0 mm i diameter). PAN-kärlsåren är segmentala och tenderar att involvera bifurcationer och grenar av artärer. Under det akuta skedet av sjukdomen infiltrerar polymorfonukleära leukocyter alla lager av kärlväggen.
Mononukleära celler är dominerande under det subakutiva steget. Under det kroniska scenet orsakar fibroidekros av kärlen trombos och vävnadsinfarkt. Aneurysmala dilatationer av de involverade artärerna, så stora som 1 cm i storlek, är karakteristiska funn av PAN.
Epidemiologin och patogenesen av PAN är okända. Experimentell immunkomplex sjukdom hos djur kan orsaka arterit, som liknar PAN. Immunofluorescensstudier under den akuta fasen av sjukdomen hos människor visar immunoglobulin och komplementavsättningar i kärlväggarna, vilket överensstämmer med en immunkomplexmedierad inflammation.
Hos patienter med cirkulerande HBsAg finns avsättningar av HBsAg ensam eller HBsAg tillsammans med anti-hepatit B immunoglobulin eller komplement i kärlväggarna. CD4 + T-celler och makrofager är närvarande i perivaskulära infiltrat. Förhållandet mellan män och kvinnor i PAN är 1, 6: 1. Sjukdomen uppträder vanligen hos vuxna i åldern 40 till 60 år.
Kliniska egenskaper:
Generellt är PAN en systemisk sjukdom och påverkar många organ. Följaktligen har patienten massor av symtom relaterade till organs involvering. PAN-patienter presenterar vanligtvis med icke-specifika tecken och symtom som feber, svaghet, huvudvärk, buksmärta, viktminskning och illamående. Vaskulära ocklusioner orsakar flera infarkt i organen och leder till organsvikt. Mikroaneurysmer i kärlväggen kan bryta och blöda.
jag. Njur:
Njurar drabbas av 60 procent av PAN-patienterna och njursvikt är den vanligaste dödsorsaken hos PAN-patienter. Reninberoende renovaskulär hypertoni förekommer hos 50 procent av patienterna.
ii. Hjärt:
60 procent av patienterna har hjärtintag (hjärtsvikt, hjärtblod och hjärtinfarkt).
III. Lung:
Lungintag kan orsaka astma, bronkit, pneumonit eller pleuritisk.
iv. GI-system:
Vaskulit i mag-tarmkanalen manifesteras av buksmärta, pankreatit, hepatit, leverinfarkt, cholecystit, tarmsjukemi och gastrointestinal blödning eller perforering.
v. CNS:
Övergående symtom på cerebral ischemi inklusive monokulär blindhet är en vanlig presentation av PAN-patienter. Stroker kan uppstå på grund av infarkt eller ruptur av mikroaneurysmer med blödning. 60 procent av patienterna utvecklar perifer neuropati. De kan ha mononeurit multiplex eller distal polyneuropati.
vi. Hud:
50 procent av PAN-patienter utvecklar hud manifestationer som livido reticularis, digitalt infarkt, palpable purpura och subkutan noduler.
vii. Öga:
Retinal vaskulit, retinal detachment och sclerit kan förekomma.
viii. urogenitala:
PAN-patienter kan utveckla smärta över testikel- eller äggstocksområdet.
Laboratorieundersökningar:
Med tanke på de ospecifika manifestationerna och mängden presentationer kan PAN vara svårt att diagnostisera. När PAN misstänks hos en individ, avslöjar angiografi och biopsi hos det drabbade organet ofta den grundläggande defekten.
jag. Perinucleära antineutrofi cytoplasmatiska antikroppar (p-ANCAs) återfinns hos 10 procent av PAN-patienter. P-ANCA är dock inte diagnostiska för PAN.
ii. ESR och CRP är upptagna.
III. PAN-patienter med njursvikt kan ha hematuri eller proteinuri och förhöjda serumkreatinin- och ureahalter.
iv. Hos HBsAg-positiva patienter uppkommer alkaliska fosfatas och serumtransaminaser.
v. Förhöjt serumamylas och lipas föreslår pankreatit.
vi. Diagnos av PAN är gjord genom biopsi eller angiografisk bekräftelse av medelstor kärlvaskulit. Biopsier av hud, muskel, sural nerv eller testikel är till hjälp vid diagnos.
vii. Viscerala angiogram är positiva hos 70 procent av PAN-patienterna. De angiografiska fynden är: förlust av den fina arborialiseringen av den viskulära vasculaturen, "korkskruvning" och vägg-oregelbundenhet hos de involverade kärlen och mikroaneurysmer av medelstora arterier. Angiogram av celiacax, överlägsen mesenterisk artär och njurartärer är vanligtvis föredragna. Behandling kan lösa de angiografiska abnormiteterna.
PAN är en potentiellt dödlig sjukdom och bör behandlas aggressivt före utveckling av irreversibla organskador. Orala kortikosteroider är ankaret för behandling av PAN. Cytotoxiska läkemedel används också vid behandling av PAN. Plasmaferes är användbar.
När den lämnas obehandlad, är 5 års överlevnad av PAN 13 procent och nästan hälften av patienterna dör under de första 3 månaderna av sjukdomsuppkomsten. Kortikosteroider förbättrar 5 års överlevnad till 50-60 procent. 5 års överlevnadshastighet kan öka upp till 80 procent när steroider kombineras med andra immunsuppressiva läkemedel. Nyresvikt är den vanligaste dödsorsaken i PAN.
Cogans syndrom:
Cogans syndrom är ett sällsynt syndrom av dövhet och keratit åtföljd (i upp till 72% fall) av en systemisk nekrotiserande vaskulit som inte kan skilja sig från PAN. Vaskuliten kan vara florid. Stora blodkärl, speciellt aorta och koronär kärl är involverade. Utlösaren av sjukdomen är okänd. Steroider med hög dos och cyklofosfamid eller cyklosporin används för att behandla sjukdomen.
Kawasakis sjukdom:
Kawasaki sjukdom (KD) eller Kawasaki syndrom eller mukokutant lymfkörtelsyndrom är en systemisk vaskulit med okänd etiologi som påverkar blodkärlen i små och medelstora storlekar, i synnerhet kransartärerna. KD kallas ibland som infantil PAN. Förekomsten av KD är högst i japanska avkomlingar. 80 procent av KD förekommer hos barn under 4 år. KD är sällsynt hos barn äldre än 14 år. KD beskrivs först i Japan 1967 av Dr Tomisaku Kawasaki.
Etiologin hos KD är okänd. Likheter mellan KD och toxic shock shock syndrom (TSS) har noterats och vissa tror att TSS och KD är olika manifestationer av samma sjukdom. Feber är det första tecknet hos många patienter, som i allmänhet har ett vaxande och avtagande mönster och varar i cirka 11 dagar i genomsnitt. Bortsett från feber behövs någon 4 av följande 5 kriterier för att diagnostisera KD.
1. Förändringar av de perifera extremiteterna, som inbegriper inledande rodnad eller ödem i handflatorna och sålarna, följt av membranaktig desquamation av finger- och tånspetsarna eller tvärgående spår över naglarna och tånaglarna (Beau linjer).
2. En polymorf, främst trunkal exantem.
3. Orofaryngeala förändringar inklusive erytem, fläckbildning och skorpning av läpparna, jordgubbstunga och diffus mukosal injektion av orpharynx.
4. Bilateral, nonxudativ, smärtfri bulbar konjunktivalinjektion.
5. Akut nonpurulent cervikal lymfadenopati med lymfkörtel diameter större än 1, 5 mm.
Det finns tre faser av sjukdomen, akut, subakut och konvalescent. Den akuta fasen av sjukdomen varar i 1-2 veckor, kännetecknad av en långvarig förhöjd temperatur. Temperaturen svarar ofta dåligt på antipyretika.
Under tredje och fjärde veckan börjar många symptom inklusive feber och utslag att lösa (subakutfas). Komplet upplösning sker vanligtvis inom 3 månader efter presentationen (ombyggnadsfas). Elektrokardiogram och ekkokardiogram behövs för att detektera koronar vaskulit. Hjärtatillverkning förekommer hos en tredjedel av patienterna. Patienter kan utveckla perikardit, kranskärl eller ventrikulär aneurysmbildning, hjärtinfarkt eller kongestivt hjärtsvikt. Döden uppträder hos 3 procent av patienterna, vanligtvis från koronar vaskulit.
jag. ESR och CRP är förhöjda.
ii. Antiendotelcellantikroppar, ANCA och cirkulerande immunkomplex är detekterbara.
III. De histologiska egenskaperna hos vaskulära lesioner är identiska med de hos PAN.
Högdos intravenös IVIg och aspirin är den huvudsakliga behandlingen av KD.
Wegeners Granulomatosis:
Wegeners granulomatos (WG) är en nekrotiserande granulomatös vaskulit som påverkar de små och medelstora arterierna och venerna. WG påverkar främst övre luftvägarna, lungparenchyma och njurar. Båda könen påverkas och toppincidensen av WG uppträder under fjärde och femte decennierna.
Etiologin hos WG är inte känd. Eftersom övre andningsorganen och lungorna påverkas, föreslås att vissa inandade antigener eller patogener kan spela en roll vid utvecklingen av WG. Förekomsten av c-ANCA i 90 procent av WG-patienter och responsen på immunosuppressiv behandling tyder på att WG är en autoimmun sjukdom.
Kliniska egenskaper:
WG-sjukdom kan förekomma som en akut, livshotande sjukdom eller som en kronisk indolent inflammatorisk sjukdom. Konstitutionella symptom som feber, viktminskning, myalgi och anorexi kan föregå de typiska manifestationerna av WG.
Luftvägar:
WG-sjukdom är misstänkt när patienter upplever symptom på övre luftvägarna (som kronisk bihåleinflammation, nässsår) eller symtom i nedre luftvägarna (som hemoptys, dyspné, hosta). 80 procent av WG-patienter har akut eller kronisk bihåleinflammation.
Näsbrosket är ofta utarmat vilket leder till gradvis kollaps av näsbryggan. Subglottisk stenos är mycket typisk och leder till akut presentation med stridor. WG kan orsaka lunginfiltrat, lungnoduler och lungblödning. Endo-bronchial inflammation kan leda till obstruktiv lungsjukdom. Interstial involvering kan leda till restriktiv lungsjukdom med andningsinsufficiens. Hilar eller mediastinala massor kan förekomma.
Öra:
Chondrit av det yttre örat, otit externt, granulomata i trumfembranet, otitis media, svimning och hörselnedsättning (ledande eller sensorisk) kan uppstå.
Njur:
80 procent av WG-patienter har glomerulonefrit och många utvecklar njurinsufficiens.
muskuloskeletala:
Cirka 70 procent av WG-patienterna klagar över myalgi och artralgi och vissa utvecklar nonerosiv artrit.
Hud:
Hudskador som palpabel purpura, kutana sår, pyoderma gangrenosum och Raynauds fenomen är vanliga i WG.
Nervsystem:
Inblandning i perifer nervsystemet orsakar mononeurit-multiplex och perifer symmetrisk polyneuropati. Central nervsystemet involverar kranial neuropati, infarkt, subdural eller subaraknoid blödning, anfall eller diffus cerebritis.
Gastrointestinala system:
Buksmärtor, diarré, tarmblödning (på grund av tarmsår) är gastrointestinala manifestationer av WG. Tarminfarkt och tarmperforering kan uppstå.
Hjärt:
10 procent av WG-patienterna har kardiala manifestationer som perikardit, kranskärlsartit, ledningsfel och kardiomyopati.
urogenitala:
Uretral obstruktion, uretrit, orchitis och epididimit kan förekomma i WG. Nekrotiserande vaskulit i blåsan kan resultera i hemorragisk cystit.
Laboratorieundersökningar:
jag. 95 procent av WG-patienter har detekterbara ANCA i deras cirkulation; varav 85 procent har c-ANCA och 10 procent har p-ANCA. Anti-neutrofil elastiska antikroppar har också detekterats. ANCA-titrar korrelerar inte med sjukdomsaktivitet. En stigande ANCA-titer kan dock ge ett återfall.
ii. 50 procent WG-patienter är positiva för RF.
III. Karakteristisk bröströntgen som visar lunginfiltrationer och knölar.
iv. Njurar: Urinsediment med RBC och RBC-gjutningar uppträder vid njurinblandning av WG. Ökad serumkreatininivå försämrar njursvikt.
v. Kreatininkinas kan förhöjas i fall med myopati.
vi. Biopsi: Positiv bekräftelse av WG görs genom transbronchial lungbiopsi eller öppen lungbiopsi. Öppen lungbiopsi ger de högsta positiva resultaten. Biopsi av nässlemhinnan visar mer särdrag hos WG. Sural nervbiopsi kan visa vasculit som påverkar små arterioler med icke-fallande granulomer som påverkar små artärer. Njurbiopsi är sällan definitiv.
vii. ESR och CRP är upptagna och dessa test kan användas för att övervaka responsen på terapi. Dessa tester kan emellertid inte förutsäga förekomst av återfall.
viii. Hos patienter med neurologiska manifestationer hjälper elektromyografi, nervledningsstudier, CSF-analys och MR-undersökningar lokaliseringen av lesionen.
ix. Det har föreslagits att indium-märkt leukocyt-avsökning är användbar för att definiera ställena för sjukdomsaktivitet.
Tecken och symtom på inflammation i övre och nedre luftvägarna, glomerulonefrit och positivt c-ANCA-resultat tyder på att sjukdomen är WG. Den slutliga diagnosen av WG bygger dock på de histologiska egenskaperna hos vaskulit, vävnadsnekros och granulom i biopsiprov.
Olika immunundertryckande regimer finns tillgängliga för behandling av WG-patienter. Tidigare WG var en dödlig sjukdom inom några månader efter diagnosen. Steroider och cyklofosfamid har förändrat scenariot. Dessa läkemedel styr effektivt sjukdomen. Utmärkt remission uppnås hos cirka 70 procent av patienterna, men tyvärr är återfallet vanligt.
Cirka 75 procent av patienterna har ihållande sjuklighet, inklusive kronisk njursvikt, nasala deformiteter, trakealstenos, kronisk bihåleinflammation och hörselnedsättning. Co-trimaxazol kan ha en sjukdomsmodifierande effekt, även om det är osäkert.
Patienterna ska få co-trimaxazol som en profylax mot Pneumocystis carinii-lunginflammation, vilket kan uppstå på grund av immunosuppression. Ciklosporin A kan vara effektivt i kombination med steroider. Högdos IVIg har föreslagits vara användbar. Anti-CD52 (Campath IH) med eller utan anti-CD4 monoklonala antikroppar eller kim-IgG-CD4-chimär molekyl är under experimentella försök. De negativa biverkningarna av långvarig användning av immunsuppressiva läkemedel bidrar till sjukligheten.
Nittiotal patienter svarar på terapi med en 5 års överlevnad som överstiger 80 procent. Men återfall är mycket vanliga när behandlingen avbryts.
Churg-Strauss syndrom:
Churg-Strauss syndrom (CSS) eller allergisk granulomatös angit är ett sällsynt syndrom som drabbar arterier och ådror från små till medelstora arter. År 1951 beskrivs syndromet av Churg och Strauss hos 13 patienter som hade astma, eosinofili, granulomatös inflammation, nekrotiserande systemisk vaskulit och nekrotiserande glomerulonefrit.
CSS är en granulomatös liten till medelstor kärlvaskulit. Etikologin hos CSS är okänd. Hypergammaglobulinemi, närvaro av reumatoid faktor (RF), närvaro av ANCA och ökad nivå av IgE föreslår dock ett autoimmunt fenomen i CSS.
CSS, Wegeners granulomatos och mikroskopisk polyangit är tre vaskulitissjukdomar, vilka är associerade med antineutrofil cytoplasmiska antikroppar (ANCA).
American College of Rheumatology (ACR) har föreslagit 6 kriterier för CSS-klassificering:
1. Astma
2. Perifer eosinofili på mer än 10 procent.
3. Para nasal bihåleinflammation.
4. Lunginfiltrat (kan vara övergående).
5. Histologiskt bevis på vaskulit med extravaskulär eosinofiler.
6. Polyneurpathy eller mononeurit multiplex.
CSS har tre faser:
Allergisk rinit och astma, infiltration av eosinofili infiltration (som eosinofili lunginflammation eller eosinofili gastroenterit) och systemisk vasculit i små och medelstora kärl med granulomatisk infiltration. Vaskulitfasen utvecklas vanligen inom 3 år efter astmas början, även om det kan vara försenat i flera årtionden.
Kliniska egenskaper:
Konstitutionella symptom som feber, viktminskning, myalgi och artralgi är vanliga hos CSS-patienter. CSS-patienter har i allmänhet en lång historia av problem med övre luftvägarna (som allergisk rinit, paras nässinus och näspolyppar), astma och eosinofili före starten av fullblåst CSS.
jag. Anemi (97% av patienterna) är den vanligaste manifestationen.
ii. Allergisk rinit, para nasal bihåleinflammation och näspolyper är de vanligaste manifestationerna i övre luftvägarna.
III. CSS-patienter kan drabbas av astma och hemoptys.
iv. Nästan hälften av CSS-patienterna har hud manifestationer (såsom purpura, subkutan noduler, urtikariautslag, nekrotisk bullae, digital ischemi).
v. Kärlvasculit med liten kärl och myokardial granulom är de främsta orsakerna till sjuklighet och mortalitet. Patienterna kan drabbas av hjärtsvikt, myokardit, myokardinfarkt, restriktiv kardiomyopati och perikardit.
vi. Mononeurit multiplex eller symmetrisk polyneuropati förekommer hos 77 procent av patienterna. Ischemisk optisk neurit är en annan manifestation av CSS.
vii. 50 procent av CSS-patienterna har fokal segmentell glomerulonefrit. Tecken på uremi och njursvikt kan utvecklas.
viii. Tarmvaskulit kan orsaka buksmärta, diarré, gastroenterit och gastrointestinal blödning. Tarmsjukemi och perforering av tarm kan förekomma.
Laboratorieundersökningar:
jag. Fullständigt blodtal avslöjar funktioner av anemi och eosinofili. Perifer eosinofil når ungefär 1000 i cirka 80 procent av patienterna.
ii. Eosinofilkationisk protein (ECP) -nivå ökar i aktiv sjukdom. ECP är neurotoxisk och kan därför vara ansvarig för några av de neurologiska problemen. Mätning av ECP kan vara till hjälp vid övervakning av sjukdomen.
III. ESR och CRP är förhöjda.
iv. Hypergammaglobulinemi och förhöjd serum IgE-nivå. Låg titer av reumatoid faktor.
v. Perinuclear ANCA (p-ANCA) är positiv hos 70 procent av CSS-patienter. C-ANCA är positiv hos 10 procent av patienterna.
vi. Proteinuri, mikroskopisk hematuri och RBC-gjutningar i urinen föreslår njursamverkan. Förhöjt serumkreatinin och urea föreslår njurinsufficiens. Eftersom njurintegration är den mest allvarliga långsiktiga sequlaen av HSP krävs upprepad urinanalys för att övervaka sjukdomsprogressionen och upplösningen. Proteinuri och hematuri är de vanligaste abnormiteterna.
vii. Imagingstudier avslöjar lungopacitet hos 26-77 procent av patienterna. Lunginfiltrering kan vara övergående.
viii. Eosinofili i bronchoalveolär lavage förekommer hos 33 procent av patienterna. Pleural effusion ses 5-30 procent av patienterna och kan vara eosinofil.
ix. Om organs involvering är närvarande är biopsi från det drabbade organet mer användbart för att komma fram till en diagnos. Annars är sural nervbiopsi den mest genomförbara proceduren. Lunghistologi visar små nekrotiserande granulomer, såväl som nekrotiserande vaskulit hos små artärer och venoler. Granulom består av en central eosinofil kärna omgiven av makrofager och epithelioida jätteceller.
Kortikosteroider används för att behandla CSS. Hos patienter som har dåligt svar på steroider eller patienter som inte kan tolerera steroider, används cytotoxiska läkemedel som cyklofosfamid eller azatioprin.
Myokardit och myokardinfarkt sekundär för kranskärlssår är de främsta orsakerna till sjuklighet och mortalitet. Med behandling är 1 års överlevnad 90 procent och 5 års överlevnad är 62 procent.
Mikroskopisk polyangit:
Mikroskopisk polyangit (MPA) är en systemisk inflammation i små blodkärl (arterioler, venules, kapillärer). MPA erkändes initialt som en mikroskopisk form av polyarterit nodosa (PAN). 1994 års internationella konsensuskonferens i Chapel Hill, North Carolina har utmärkt MPA från PAN. I PAN är vaskulit av små kärl inklusive arterioler, kapillärer och venules frånvarande. Vasculit av små kärl förekommer i MPA.
MPA kännetecknas av pauciimmun, nekrotiserande liten kärlvaskulit utan kliniskt eller patologiskt bevis på nekrotiserande granulomatös inflammation. Sjukdomsåldern är cirka 50 år. Manspersoner påverkas något mer än kvinnorna. Njurinsufficiens och lungsjukdomar är de främsta orsakerna till sjuklighet och dödlighet.
Kliniska egenskaper:
Liksom andra vaskulitsyndrom finns konstitutionella symptom som feber, myalgi, viktminskning och anorexi i MPA.
jag. Njur:
Rapidly progressive crescentric glomeruloneephritis förekommer hos nästan alla MPA-patienter. Det finns inga immunförsvar i njurarna. Utan behandling utvecklas njursvikt snabbt.
ii. Lung:
Hos 50 procent av MPA-patienter påverkas lungorna. Lungkapillarit leder till blödning och hemoptys. Interstitiell fibros uppträder senare.
III. muskuloskeletala:
Myalgi, artralgi och icke-deformerande artrit ses hos 75 procent av patienterna.
iv. Hud:
Hudutslag, palpabel purpura, livido reticularis, urtikaria och sår i huden ses.
v. gastrointestinal:
Buksmärtor, diarré och hepatosplenomegali förekommer i MPA.
vi. Öga:
Episklerit, uveit, retinalblödning, okulär smärta och nedsatt syn förekommer i MPA.
vii. Nervsystem:
Mononeuritmultiplex förekommer hos 57 procent av MPA-patienter. Beslag uppträder hos 11 procent av MPA-patienter.
viii. Hjärt:
Perikardit, bröstsmärta och hjärtsvikt.
Laboratorieundersökningar:
jag. 80 procent av MPA-patienterna är positiva för ANCA. 80 procent av dessa patienter är positiva för p-ANCA och resterande 20 procent är positiva för c-ANCA.
ii. ESR och CRP är förhöjda. RF är positiv i över hälften av MPA-patienterna. Antinucleära antikroppar är positiva hos en tredjedel av patienterna.
III. Renal involvering leder till hematuri, proteinuri, urinmedelssediment och slutligen till renhetssvikt.
iv. Histologi:
Den slutgiltiga diagnosen av MPA beror på den histologiska observationen av pauciimmun, nekrotiserande vaskulit hos små kärl. Hud, lunga, njure och sural nerv är de vanliga platserna för biopsi.
v. Angiografi av de mesenteriska kärlen visar inte mikroaneurysmer, vilket skiljer MPA från PAN.
Immunsuppressiva läkemedel används för att behandla MPA. 5 års överlevnadshastighet hos patienter med fulminant MPA är 65 procent. Återfall är vanliga om behandlingen avbryts.
Henoch-Schonlein Purpura:
Henoch-Schonlein purpura (HSP) är en IgA-medierad vasculit med små kärl som huvudsakligen påverkar barn men ses också hos vuxna. En till två procent av patienterna utvecklar njursvikt. Heberden beskrev först sjukdomen 1801 i ett 5 år gammalt barn med buksmärtor, hematuri och purpura i benen. År 1837 beskrev Johann-Schonlein ett syndrom av purpura i samband med ledsmärta och urinfällningar hos barn.
Edward Henoch ytterligare associerad buksmärta och njursamverkan med syndromet. Etikologin hos HSP är inte känd. Hos 50 procent av barnen förekommer infektion i övre luftvägarna före starten av HSP. Andra faktorer (som droger, malignitet, livsmedel, familjen Medelhavsfeber, exponering för kyla) har också associerats med utvecklingen av HSP. HSP har rapporterats efter vaccinationer mot tyfoid, mässling, gul feber och kolera.
HSP innefattar vaskulär avsättning av IgA-innehållande immunkomplex, orsaken för vilken inte är känd. IgA-innehållande immunkomplex deponering i väggarna i små kärl (artärer, kapillärer, venules) och aktivera komplementsystemet.
Det mest sannolika komplementssystemet som aktiveras i HSP verkar vara den alternativa vägen för komplementet, eftersom properdin- och C3-avsättningar i frånvaro av Clq-avlagringar ses i de flesta biopsierna. Komplement aktivering leder till frisättning av inflammatoriska mediatorer och efterföljande infiltrering av platsen av inflammatoriska celler.
Den inflammatoriska reaktionen resulterar i kärlväggnekros med samtidig trombos. Följaktligen uppträder blödning i de drabbade organen och manifesteras histologiskt som leukocytoklastisk vaskulit.
Kliniska egenskaper:
HSP-patienter presenterar ofta med låg grad av feber och illamående utöver specifika symtom. De kan presentera med purpura. Omkring 50 procent av barnen uppvisar andra symtom än purpura. Den purpuriska utbrottet föregås av artralgi eller artrit, buksmärta eller svullnad i testiklar.
jag. Skada på kärl av papillär dermis på grund av IgA och C3-avsättningar leder till kärlskada; följaktligen utvecklas extravasationer av RBC och kliniskt observerbar purpura. Hallmärket för sjukdomen är palpabel purpura som förekommer hos 100 procent av HSP-patienter. Purpura tenderar att förekomma i kroppsberoende kroppsdelar, såsom underben, skinkor, rygg och buk.
ii. Sextiofem procent av patienterna har buksmärta, som är sekundär mot vaskulärinducerad submukosal och subserosal blödning och ödem med trombos av mikrovaskulaturen i tarmen. Buksmärtor med samtidig hematochezia är det näst vanligaste symptomet. Cirka 6 procent av patienterna behöver kirurgi för komplikationer som intussusception, tarmvägg perforation eller infarkt.
III. I HSP-medierad nephritis förekommer mikroskopisk hematuri och proteinuri, som varar i dagar till veckor. Detta kan följas av mikroskopisk hematuri, som kan varas i månader till år. Vissa patienter utvecklar kroniskt njursvikt och slutstadie njursjukdom. Glomerulär mesangial deponering av IgA uppträder övervägande; men IgG, IgM, C3 och properdin-avsättningar ses också.
iv. Lungblödning är sällsynt men en dödlig komplikation av HSP.
Laboratorieundersökningar:
jag. Urin
Proteinuri och mikroskopisk hematuri är vanliga abnormiteter i urinen. Eftersom njurinblandning kan förekomma i HSP bör urinanalys utföras månadsvis i flera månader efter presentation.
ii. ESR och CRP är förhöjda.
III. Direkt immunofluorescensmikroskopi (DIFM):
DIFM av biopsiprover visar dominans av IgA-deponering i kärlväggarna hos drabbade vävnader. Perilionell hud intill den kutana skadorna kan också visa IgA-avsättningar. Njurbiopsiprover visar mesangiala, granulära IgA-avsättningar, ofta med C3, IgG eller IgM.
iv. Histologi:
Leukocytoklastisk vaskulit är det dominerande resultatet i drabbade vävnader. Hudbiopsi demonstrerar fibrinoidnekros av arteriolära och venulära väggar i ytlig dermis, med neutrofilt infiltration av väggarna och perivaskulära regioner. Mukosalbiopsi från drabbade mag-tarmkanalen visar identiska histologiska egenskaper som ses i huden. Renalbiopsi visar ett spektrum av glomerulär sjukdom från minimal förändring till allvarlig kressentrisk glomerulonephritis.
v. Förhöjda nivåer av serum IgA och cirkulerande immunkomplex innehållande IgA kan detekteras. IgA-reumatoidfaktor har rapporterats hos HSP-patienter. HSP är vanligtvis en självbegränsande sjukdom och behandling är stödjande för att säkerställa adekvat hydrering och ersättning av blod om det behövs. Användningen av kortikosteroider i HSP är kontroversiell. Aspirin bör undvikas eftersom det kommer att förvärra tarmblödningen. Icke-steroida antiinflammatoriska läkemedel kan användas för att behandla artralgi. Prognosen är generellt utmärkt. Kroniskt njursvikt kan emellertid inträffa hos 2-5 procent av patienterna.
IgA nefropati (Berger's sjukdom) är noga nära relaterad till HSP. IgA nefropati kan vara HSP med njursjukdom men ingen utslag. IgA nefropati föregås också av infektioner i övre luftvägarna, det kan förekomma tarmintag och artralgi. Även IgA-nefropati associeras med IgA-immunkomplex och IgA-reumatoid faktor.
Essential Cryoglobulinemic Vasculitis:
Viktig cryoglobulinemi är associerad med feber, hudabcesser, artralgier och Raynaud-syndrom, Sjogren-syndrom eller båda. Glomerulonefrit och perifer neuropati ses ofta. Viktig kryoglobulinemi klassificeras i tre typer, typ I, typ II och typ III.
Typ II (väsentlig blandad kryoglobulinemi) är associerad med purpura, Raynauds fenomen, artralgi, perifer neuropati, Sjogrens syndrom, glomerulonephritis och leversjukdom. There is usually IgMK paraproteinemia and evidence for a low-grade lymphoproliferative disease. The disease is very common in northern Italy and associated with chronic Hepatitis C (HCV) infection. Other chronic bacteremic infections such as shunt nephritis and low-grade endocarditis may also lead to cryoglobulin formation.
Leucocytoclastic Vasculitis:
Leucocytoclastic vasculitis (LCV) is a histopathologic term commonly used to denote a small-vessel vasculitis. There are many causes for this condition, but a cause is not found in as many as 50 percent of patients. LCV may be localized to the skin or it may manifest in other organs. The joints, gastrointestinal tract, and the kidneys are commonly affected. The disease may be acute or chronic.
The exact causes of LCV are not known. In the past, circulating immune complexes were believed to be responsible for LCV. Autoantibodies (such as ANCA) and local factors that involve endothelial cell adhesion molecules may play important roles. LCV appears to be reported more often in the white population.
Patients with vasculitis of skin may complain of itching, burning, or pain or the lesion may be asymptomatic. Vasculitis may occur in the absence of any systemic disease or it may occur in conjunction with collagen vascular disorders, paraproteinemia, certain food, medications (such as antibiotics, non-steroidal antiinflammatory drugs, and diuretics), infections (such as Hepatitis B virus. Hepatitis C virus), or, rarely, malignancy.
jag. Palpable purpura is the most common manifestation of cutaneous vasculitis.
ii. The utricarial lesions tend to be different from routine urticaria. The vasculitis urticaria tends to be of longer duration (often longer than 24 hours) and tend to resolve with some residual pigmentation or echymosis. The patients complain more of burning rather than itching.
Skin biopsy reveals vascular and perivascular infiltration of polymorphonuclear leukocytes with formation of nuclear dust (leukocytoclasis), extravasation of erythrocytes, and fibrinoid necrosis of the vessel walls. Immunofluorescent staining may reveal immunoglobulins and complement component deposits on the skin basement membrane.
Chronic cutaneous disease may involve ulceration or painful bouts of purpura. The prognosis of LCV is generally good. However, mortality is possible when lungs, kidney, heart, or CNS are involved.
Thromboangitis Obliterans:
Thromboangitis obliterans (TA) or Buerger's disease (BD) is a nonatherosclerotic, segmental, inflammatory, vasoocclusive disease that affects the small-and medium- sized arteries and veins in the upper extremities and lower extremities. The etiology and pathogenesis of TA are not known. In the early 1900s, Leo Buerger published a detailed description of the disease in which he referred to the clinical presentation of TA as 'presenile spontaneous gangrene'.
TA is closely linked to the use of tobacco. Exposure to tobacco is essential for the initiation and progression of the disease. The association of tobacco exposure is supported by the observation that TA is more common in countries with heavy tobacco use and the disease is perhaps more common among natives of Bangladesh, who smoke a specific type of cigarettes, made from tobacco, called 'bidi'. However, a few cases of TA in nonsmoking individuals have been described and it is attributed to the use of tobacco chewing. Natives of India, Korea, Japan, and Israeli Jews of Ashkenazi descent have the highest incidence of TA.
The mechanism of development of TA in unknown. The following observations suggest the possibility of an immunologic phenomenon that leads to vasodysfunction and inflammatory thrombi formation.
jag. TA patients show hypersensitivity to intradermally injected tobacco extracts.
ii. TA patients have elevated serum anti-endothelial cell antibody titers.
Most patients with TA are aged between 20 to 45 years. TA is more common in men and it may be due to an increased incidence of tobacco smoking by men.
70-80 percent of TA patients present with distal, ischemic, resting pain and / or ischemic ulcerations on the toes, feet, or fingers. Patients may present with claudication of the feet, legs, hands, or arms and often describe the Raynaud phenomenon of sensitivity of the hands and fingers to cold. Patients may develop painful ulcerations and / or frank gangrene of the digits. Impaired distal pulses in the presence of normal proximal pulses are usually found in TA patients.
Classic TA affects vessels of the extremities. However, a few cases of aortic, cerebral, coronary, iliac, mesenteric, pulmonary, and renal thromboangitis obliterans have been reported.
jag. Angiography:
Non atherosclerotic, segmental occlusive lesions of the small-and medium-size vessels (such as digital, palmar, plantar, tibial, peroneal, radial, and ulnar arteries) with formation of distinctive small vessel collaterals around areas of occlusion known as “corkscrew collaterals” are the hallmark of TA. However, such lesions can also be observed in scleroderma, SLE, rheumatoid vasculitis, mixed connective tissue disorders (MCTD), and anti- phospholipid antibody syndrome.
ii. Histologi:
In the acute stage, highly cellular, segmental, occlusive, inflammatory thrombi, with minimal inflammation in the walls of affected blood vessels are observed. In the subacute stage there is progressive organization of intraluminal thrombosis.
The end stage phase of the disease is characterized by mature thrombus and vascular fibrosis. In all the 3 stages, the integrity of the normal structure of the vessel wall, including the internal elastic lamina is maintained. This feature distinguishes TA from arteriosclerosis and from other types of systemic vasculitis, in which disruption of the internal elastic lamina and the media may be extensive.
Death from TA is rare. But in patients who continue to smoke, 43 percent require one or more amputations in 7.6 years. Absolute discontinuation of tobacco use is essential to prevent the progression of the disease. Intravenous iloprost (a prostaglandin analogue) therapy shows symptomatic improvement and reduces the amputation rate among TA patients.
Vasculitis Secondary to Other Diseases:
Vasculitis may occur secondary to other disease process, some of which are listed below.
infektioner:
Bacteria: Streptococcus, spirochetes (Treponema pallidum, Borrelia), mycobacteria, rickettsia.
Virus:
Varicella zoster virus, cytomegalovirus. Hepatitis A virus. Hepatitis B virus. Hepatitis C virus. Influenza virus.
Svamp:
Malignancy:
Hairy-cell leukemia, lymphoma, acute myeloid leukemia, with and without cryofibrinogens.
Narkotika:
Oral contraceptives, sulphonamides, penicillin's, thiazides, aspirin, cocaine, amphetamines, LSD.
Secondary to other autoimmune diseases:
Primary biliary cirrhosis. Good pasture's syndrome, SLE, rheumatoid arthritis, systemic sclerosis, Sjogren's syndrome, polymyositis / dermatomyositis, hypocomplementemic urticaria, relapsing polychondritis.
Secondary to inflammatory bowel disease, ulcerative colitis, Crohn's disease.
Hypocomplementemic Vasculitis:
The hypocomplementemic vasculitis is characterized by urticaria with hypocomplementemia. Angioedema may also occur. Unlike allergic diseases, the hypocomplementemic urticaria may be prolonged, lasting up to 72 hours and the skin lesions leave residual staining of the skin.
40 percent of patients have renal involvement with granular IgG deposits along the glomerular basement membrane, and obstructive lung disease has also been documented. The CH 100 is low. Clq, C2, and C4 levels are low. An autoantibody to the Clq component is suggested to activate the classic complement pathway.
A similar syndrome may occur also occur with C3- nephritic factor.
Steroids or dapsone (impairs chemotaxis and lysosomal activity of neutrophils and neutrophil adherence) are used to treat the condition.
Certain conditions such as cholesterol embolus, myxoma embolus, and ergotism mimics vasculitis.